
Mental illnesses can affect our brain in more ways than just mood.
Depression is one of the most widely studied psychiatric disorders in the world. There’s plenty of focus on its outward signs from mental health resources to depictions in pop culture. A few words might spring to mind — sad, irritable or lethargic. But depression is far from a bad case of the blues or just an imbalance of chemicals. It can impact the brain itself in a variety of ways with visible, pathological ramifications.
Depression can cause the brain to shrink
Studies have demonstrated that depression can cause certain brain regions to physically change in size. This includes the hippocampus, amygdala, thalamus and other parts of the prefrontal cortex [1, 2]. A primary area of interest is the hippocampus—an S-shaped structure packed deep in the brain that regulates learning, memory and emotion. A 2013 study in postmortem tissues found that patients suffering from depression for a longer time had smaller hippocampuses, with its total volume decreasing by almost 1000 mm3 over 30 years (Figure 1) [3]. Interestingly, the density of pyramidal neurons in the CA1 region of the hippocampal circuit—central to projecting signals and many excitatory processes—increased over the same period. However, it was associated with a shrinkage of the soma (the core of neurons containing the nucleus) [3–5]. Elevated levels of stress hormones such as glucocorticoids associated with depression are thought to be the primary factor behind this shrinkage. These hormones reduce neuron formation and could even lead to cell death, causing a loss of hippocampal function [6]. Considering its role, outward signs of hippocampal atrophy seen in depression may manifest as difficulty concentrating, memory deficits and other cognitive impairments [7].
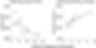
Figure 1. Changes in hippocampal volume and CA1 pyramidal neuron density as a function of depression duration. [3]
While this may sound unnerving, contrary to popular belief, the brain has the remarkable ability to create new neurons and connections. Antidepressants have been shown to promote the body’s secretion of peptides called neurotrophic factors which increase neuron maturation and proliferation. In cases where shrinkage of certain brain regions is observed, these factors likely have a restorative effect [6]. However, medication alone is not a silver bullet, and hippocampal atrophy might not mean that someone must be depressed. For instance, a 2005 study found that patients who had recovered from clinical depression and who were not on medication still had reduced hippocampal volumes compared to healthy subjects (Figure 2) [8]. So while a reduction in hippocampus size is observed in depression, there are still questions about their exact correlation and where antidepressants fit into the picture.

Figure 2. Total, posterior and anterior hippocampal volumes of patients who had remitted from depression and were unmedicated at the time of the study versus healthy subjects. Mean time since remission: 30.5 months [8].
So how does stress lead to neuronal damage?
As noted, chronic stress in depression is a primary cause of neuronal atrophy. To understand how this happens, we must examine the link between stress and the inflammatory response. When we get cuts and scrapes, we often see them swell up and redden. That’s because inflammatory mediators called cytokines promote blood flow and stimulate the immune system to fight infection. Although depression isn’t exactly an external injury, psychological stress also stimulates cytokine activation [9, 10].
This is a problem because excess cytokine production in the brain during depression can interfere with the normal functioning of many neurotransmitter systems [11]. For example, elevated levels of the cytokine interferon (IFN) can induce release of more inflammatory mediators. This activates enzymes to break down tryptophan—the precursor needed to form serotonin, which has a key role in mood regulation (Figure 3) [9, 12]. Increased tryptophan metabolism therefore contributes to the chemical imbalance involved in depression, expressing itself as low mood, anhedonia and irritability [13].
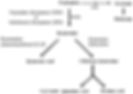
Figure 3. Major metabolic pathway of the serotonin precursor tryptophan in humans. IFN increases the activity of indoleamine dioxygenase (IDO) to increase tryptophan breakdown, thus reducing serotonin availability [12].
Disturbance of this metabolic pathway has another unpleasant effect. When tryptophan breaks down, it forms a product called quinolinic acid, which causes an excessive influx of calcium into neurons. This overstimulates many cellular signalling pathways, leading to mitochondrial dysfunction, oxidative stress and cell death (Figure 4) [14, 15]. Furthermore, a 2018 study found 29–33% greater activation of immune cells in parts of the cortex of patients who had experienced prolonged untreated depression (>10 years) [16]. An increase of cytokine production by these immune cells could speed up tryptophan metabolism and cause quinolinic acid formation. Hence, activation of the inflammatory cascade during stress is thought to be a key contributor of neuronal loss in depression.
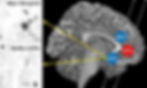
Figure 4. Illustration of the subgenual anterior cingulate cortex (sACC) in a depressed vs. healthy subject. Quinolinic acid-associated neurons are stained, showing greater prevalence in the depressed individual and association with cell death [15].
Where can we go from here?
Although the impacts of depression on the brain are concerning, it does offer a unique therapeutic window. Understanding the role of stress and inflammation in depression could let us create new and selective medicines down the road. One such drug in development reduces cytokine synthesis by blocking and preventing activation of the P2X7 receptor in the brain, thereby decreasing inflammation and neuronal injury in depression. Recent human clinical trials have displayed good tolerance and possible mood-modulating effects [17]. Therefore, as our knowledge of depression continues to advance, so too will the potential to develop better and safer therapeutics for patients in need.
References:
[1] Palazidou E. The neurobiology of depression. British Medical Bulletin. 2012;101(1):127-45.
[2] Yüksel D, Engelen J, Schuster V, Dietsche B, Konrad C, Jansen A, et al. Longitudinal brain volume changes in major depressive disorder. Journal of Neural Transmission. 2018;125(10):1433-47.
[3] Cobb JA, Simpson J, Mahajan GJ, Overholser JC, Jurjus GJ, Dieter L, et al. Hippocampal volume and total cell numbers in major depressive disorder. Journal of Psychiatric Research. 2013;47(3):299-306.
[4] Bekkers JM. Pyramidal neurons. Current biology. 2011 Dec 20;21(24):R975.
[5] Stockmeier CA, Mahajan GJ, Konick LC, Overholser JC, Jurjus GJ, Meltzer HY, Uylings HB, Friedman L, Rajkowska G. Cellular changes in the postmortem hippocampus in major depression. Biological psychiatry. 2004 Nov 1;56(9):640-50.
[6] Planchez B, Surget A, Belzung C. Adult hippocampal neurogenesis and antidepressants effects. Current Opinion in Pharmacology. 2020;50:88-95.
[7] Rahman MM, Callaghan CK, Kerskens CM, Chattarji S, O’Mara SM. Early hippocampal volume loss as a marker of eventual memory deficits caused by repeated stress. Scientific reports. 2016 Jul 4;6(1):1-5.
[8] Neumeister A, Wood S, Bonne O, Nugent AC, Luckenbaugh DA, Young T, Bain EE, Charney DS, Drevets WC. Reduced hippocampal volume in unmedicated, remitted patients with major depression versus control subjects. Biological psychiatry. 2005 Apr 15;57(8):935-7.
[9] Raison CL, Capuron L, Miller AH. Cytokines sing the blues: inflammation and the pathogenesis of depression. Trends in immunology. 2006 Jan 1;27(1):24-31.
[10] Madrigal JL, Hurtado O, Moro MA, Lizasoain I, Lorenzo P, Castrillo A, Boscá L, Leza JC. The increase in TNF-α levels is implicated in NF-κB activation and inducible nitric oxide synthase expression in brain cortex after immobilization stress. Neuropsychopharmacology. 2002 Feb;26(2):155-63.
[11] Felger JC, Lotrich FE. Inflammatory cytokines in depression: neurobiological mechanisms and therapeutic implications. Neuroscience. 2013 Aug 29;246:199-229.
[12] Höglund E, Øverli Ø, Winberg S. Tryptophan metabolic pathways and brain serotonergic activity: a comparative review. Frontiers in endocrinology. 2019 Apr 8;10:158.
[13] Capuron L, Neurauter G, Musselman DL, Lawson DH, Nemeroff CB, Fuchs D, Miller AH. Interferon-alpha–induced changes in tryptophan metabolism: relationship to depression and paroxetine treatment. Biological psychiatry. 2003 Nov 1;54(9):906-14.
[14] La Cruz VP, Carrillo-Mora P, Santamaría A. Quinolinic acid, an endogenous molecule combining excitotoxicity, oxidative stress and other toxic mechanisms. International Journal of Tryptophan Research. 2012 Jan;5:IJTR-S8158.
[15] Steiner J, Walter M, Gos T, Guillemin GJ, Bernstein HG, Sarnyai Z, Mawrin C, Brisch R, Bielau H, zu Schwabedissen LM, Bogerts B. Severe depression is associated with increased microglial quinolinic acid in subregions of the anterior cingulate gyrus: evidence for an immune-modulated glutamatergic neurotransmission?. Journal of neuroinflammation. 2011 Dec;8(1):1-9.
[16] Setiawan E, Attwells S, Wilson AA, Mizrahi R, Rusjan PM, Miler L, Xu C, Sharma S, Kish S, Houle S, Meyer JH. Association of translocator protein total distribution volume with duration of untreated major depressive disorder: a cross-sectional study. The Lancet Psychiatry. 2018 Apr 1;5(4):339-47.
[17] Recourt K, van der Aart J, Jacobs G, de Kam M, Drevets W, van Nueten L, Kanhai K, Siebenga P, Zuiker R, Ravenstijn P, Timmers M. Characterisation of the pharmacodynamic effects of the P2X7 receptor antagonist JNJ-54175446 using an oral dexamphetamine challenge model in healthy males in a randomised, double-blind, placebo-controlled, multiple ascending dose trial. Journal of Psychopharmacology. 2020 Sep;34(9):1030-42.